
 資料結構
資料結構 網路
網路 關係資料庫管理系統
關係資料庫管理系統 作業系統
作業系統 Java
Java iOS
iOS HTML
HTML CSS
CSS Android
Android Python
Python C 程式設計
C 程式設計 C++
C++ C#
C# MongoDB
MongoDB MySQL
MySQL Javascript
Javascript PHP
PHP卡納萬病
卡納萬病屬於一類稱為白質營養不良的疾病家族。白質營養不良是一種罕見、漸進性、代謝性、遺傳性疾病,可對神經系統結構(周圍神經)產生深遠影響,這些結構超出了大腦和脊髓的範圍。至少有十種不同型別的白質營養不良,每種都是由影響不同基因的不規則性引起的,並導致大腦白質中發現的不同化學物質的異常發育。神經纖維構成大腦的白質。髓鞘是由蛋白質和脂質組成的脂肪膜,可以保護許多神經纖維。髓鞘是神經纖維的保護層,也起到絕緣材料的作用,並有助於更快地傳遞訊號。不同型別的白質營養不良會影響髓鞘的不同成分,從而導致各種神經系統症狀。

什麼是卡納萬病?
像卡納萬病這樣的腦部疾病並不常見,通常由基因引起。該疾病的神經退行性特徵意味著大腦的異常會隨著時間的推移而加重。當一種關鍵物質缺失時,大腦會變得海綿狀且功能不佳。白質營養不良是一類包括卡納萬病在內的疾病。這些疾病是進行性、罕見、遺傳性疾病,在整個神經系統中以各種方式表現出來。
卡納萬病的症狀有哪些?
發育遲緩
受影響的新生兒在達到發育階段(例如,無支撐站立或坐立)方面也表現出延遲,大多數人從未獨立行走。在整個嬰兒期,需要精神和身體活動同步(運動協調退化)的技能的逐漸喪失以及發育遲緩也變得明顯。大多數患病新生兒學會咧嘴笑、咯咯笑、抬起頭部並進行社互動動。
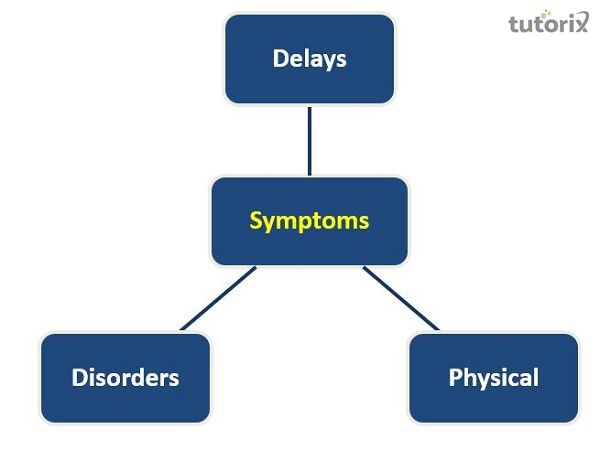
疾病
有時還會伴有癲癇發作、睡眠障礙、餵養問題、鼻腔反流和反流(胃酸迴流到食道)。眼窩(視神經末梢)的神經末梢退化也很常見於卡納萬病患兒(視神經萎縮),眼窩的神經末梢將來自眼瞼內襯的震動膜(視網膜)的脈衝傳送到大腦。視神經萎縮會導致視覺反應下降。聽力損失並不常見;因此,聽力通常不受影響。
身體
如果不治療,嬰兒的肌張力低下可能會發展為痙攣,這是一種以導致腿部緩慢、僵硬運動的非自主肌肉痙攣為特徵的疾病。去大腦僵直是一種患者的胳膊、四肢、指尖和腳趾變得僵硬且無法移動的疾病。卡納萬病是一種進行性疾病,可能導致致命後果,儘管每個人的病情發展速度和這些問題的嚴重程度可能差異很大。一些嬰兒有危及生命的困難,而另一些嬰兒則活到了成年。
卡納萬病的原因是什麼?
當受影響的個體從父母雙方各獲得一份有缺陷的基因副本時,就會出現隱性遺傳疾病。攜帶者是指攜帶該疾病但未表現出任何症狀的人,因為他們獲得了一個人類基因和一個疾病基因。每對攜帶者父母的懷孕都有 25% 的機率產生患病的孩子。每次懷孕都有 50% 的機率會像父母一樣產生攜帶者孩子。孩子有 25% 的機率從父母雙方遺傳該性狀的正常基因。男女都面臨著同樣的威脅。
卡納萬病是如何診斷的?
嬰兒表現出卡納萬病的典型症狀會引起對診斷的懷疑。全面的臨床評估結果,包括完整的病史和一系列專門的測試,可能有助於確認診斷。Gc 光譜法是一種可以識別尿液中 NAA 水平升高的工具,可用於此類檢查。NaN3 乙醯轉移酶活性在血漿和脊髓液 (CSF) 中也升高。為了檢測天冬醯胺酶缺乏症,研究人員可以分析培養的成纖維細胞,即皮膚結締組織中發現的細胞。
此外,白細胞無法產生天冬醯胺酶。羊膜穿刺術在懷孕 16 至 18 周之間進行,透過評估正在發育的嬰兒周圍羊水中 NAA 的含量來檢測母親血液中的卡納萬病。絨毛膜絨毛取樣 (CVS) 允許產前診斷,如果父母雙方都具有已識別的 ASPA 基因突變,則透過在妊娠 10-12 周時取出一塊胎盤細胞樣本進行突變檢查。
卡納萬病如何治療?
卡納萬病的治療是根據患者的具體病情表現量身定製的。提供支援性護理可以緩解一些疼痛。物理治療和早期干預可能有利於姿勢和語言技能。如果患者吞嚥困難,可以透過鼻飼管幫助他們獲得所需的營養和液體。對於那些患有癲癇發作的人來說,可以使用抗癲癇藥物。基因治療作為治療卡納萬病患兒的潛在療法正在研究中。醫生使用基因治療將基因的功能複製引入其發育中的神經系統,以治療患有 ASPA 缺陷的兒童。正是這些基因產生了天冬醯胺酶,這是一種降解 NAA 所必需的酶。接受基因治療的兒童的症狀已顯著減輕。基因治療已被證明是一種很有希望的治療卡納萬病患者的方法,但需要進一步的研究來確認其長期安全性和有效性。
卡納萬病無法治癒,也沒有確立公認的治療方案。有效的治療和支援形式。有些人從物理治療中受益,以提高他們的運動技能,而另一些人則從教育專案中受益,以提高他們的語言能力。抗癲癇藥物用於治療癲癇發作,在患者吞嚥困難的情況下進行胃造口術,使他們難以攝入足夠的食品或液體。
結論
在嬰兒和幼兒中發現了卡納萬病的確診病例。由於缺乏代代相傳的酶,導致髓鞘(大腦中神經細胞的保護層)退化。

115 次檢視
